


Complejidad de las bases genéticas de los trastornos NBIA
RiuNet: Repositorio Institucional de la Universidad Politécnica de Valencia
JavaScript is disabled for your browser. Some features of this site may not work without it.
Buscar en RiuNet
Listar
Mi cuenta
Estadísticas
Ayuda RiuNet
Admin. UPV

Complejidad de las bases genéticas de los trastornos NBIA
Mostrar el registro sencillo del ítem
Ficheros en el ítem

| dc.contributor.advisor | Espinós Armero, Carmen Ángeles
|
es_ES |
| dc.contributor.advisor | Lupo, Vincenzo
|
es_ES |
| dc.contributor.advisor | Forment Millet, José Javier
|
es_ES |
| dc.contributor.author | Guillot Fernández, Marina
|
es_ES |
| dc.date.accessioned | 2019-10-07T11:53:02Z | |
| dc.date.available | 2019-10-07T11:53:02Z | |
| dc.date.created | 2019-09-20 | |
| dc.date.issued | 2019-10-07 | es_ES |
| dc.identifier.uri | http://hdl.handle.net/10251/127560 | |
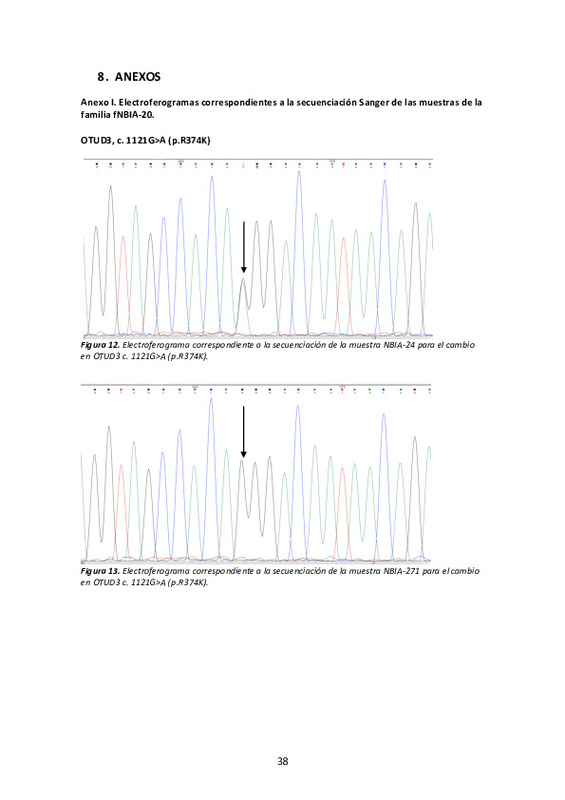
| dc.description.abstract | [ES] Las ENACH o NBIA (Neurodegeneration with Brain Iron Accumulation) comprenden un grupo heterogéneo de enfermedades hereditarias que suelen presentar depósitos de hierro en el cerebro, principalmente en los ganglios basales. Se trata de enfermedades raras con una prevalencia de 1/1.000.000. Hasta la fecha se conocen 12 genes NBIA, aunque un número relevante de pacientes permanece sin diagnóstico genético. Alrededor del 35-50% de pacientes NBIA padecen PKAN (Pantothenate Kinase-Associated Neurodegeneration, gen PANK2). La segunda forma más frecuente es PLAN (PhosphoLipase A2 Associated Neurodegeneration, gen PLA2G6) que responde a aproximadamente el 20% de los pacientes NBIA. El resto de formas son poco frecuentes. El avance de estas enfermedades es devastador e incluye dificultades progresivas en el habla, disfagia, distonía, espasticidad, parkinsonismo, acinesia pura, disfunción oculomotora, pérdida de visión y síntomas neuropsiquiátricos. Los pacientes precisan de la atención de un equipo multidisciplinario y reciben un tratamiento paliativo que se centra principalmente en reducir la distonía y la espasticidad que son altamente discapacitantes. En 2015 iniciamos un proyecto financiado por la Fundació La Marató de la TV3 coordinado por la Dra. B. Pérez-Dueñas (H.U. Vall d'Hebron) para la elaboración de una escala clínica para pacientes PKAN y el diagnóstico genético de la población española NBIA con la colaboración de más de 20 hospitales españoles. Algunos de los clínicos que forman parte de esta red, forman parte del equipo de investigación del presente proyecto. La escala clínica PKAN-DSR (PKAN Disease Rating Scale) ha sido ya validada con éxito en la evaluación clínica de nuestra serie16. Actualmente tenemos censados 134 casos NBIA o NBIA-mimic, que refiere a aquellas formas que no cursan estrictamente con el cuadro clínico característico de estos pacientes. El análisis genético de PANK2 y PLA2G6 mediante secuenciación Sanger y MLPA nos ha permitido diagnosticar el 54% de los casos. Los pacientes que resultan negativos para estos análisis son analizados mediante un panel diseñado con tecnología SureSelectXT (Agilent) para MiSeq (Illumina), que comprende 498 genes implicados en NBIA y otros trastornos del movimiento (MovDisord-498). Hemos analizado con esta metodología 38 pacientes y de momento, hemos logrado el diagnóstico genético en 19 de ellos. Los resultados genéticos han revelado que el espectro clínico es más amplio del conocido hasta ahora. Un ejemplo de todo ello es la paciente NBIA-18. Se trata de una niña de padres consanguíneos en quien hemos detectado dos cambios en homozigosis en dos genes: DLD y FBXO7. Hemos llevado a cabo estudios de expresión (western-blot), de Nonsense Mediated Decay (NMD) y de localización sub-celular en fibroblastos. Éstos han revelado que la mutación FBXO7 c.368C>G (p.S123X) conduce a la activación del mecanismo NMD y el mRNA es degradado; y que la mutación DLD c.100A>G (p.T34A) impide que la proteína alcance su localización correcta en la mitocondria. Los casos que son negativos tras su estudio mediante el panel MovDisord-498 son estudiados mediante secuenciación de exoma (WES, Whole Exome Sequencing). Está descrito que aproximadamente el 85% de las mutaciones patológicas se localizan en los exones. Por ello, pese a que la WES sólo supone la secuenciación de alrededor del 2% del genoma humano, se ha demostrado que es una herramienta coste-efectiva interesante para el diagnóstico genético. En el caso de enfermedades pediátricas del neurodesarrollo, estudios comparativos has mostrado que cuando sólo se aplica WES a la muestra del probando, la tasa de éxito ronda el 28%, mientras que si se amplía el estudio a los padres (trío) ésta aumenta un 38%. Es importante recalcar que la mayoría de casos son esporádicos y por tanto, es de esperar mutaciones de novo o bien, mutaciones con una herencia autosómica r | es_ES |
| dc.description.abstract | [EN] The disorders with NBIA (Neurodegeneration with Brain Iron Accumulation) comprise a heterogeneous group of hereditary diseases which are characterized by the presence of iron deposits in the brain, mainly in the basal ganglia. These diseases are rare, with a prevalence of 1/1.000.000. So far, 12 genes causing NBIA diseases have been described, but there is a relevant number of patients lacking a genetic diagnostic. Around 35-50% of NBIA patients suffer from PKAN (Pantothenate Kinase-Associated Neurodegeneration, PANK2 gene). The second most frequent form is PLAN (PhosphoLipase A2 Associated Neurodegeneration, PLA2G6 gene), present in approximately 20% of the NBIA patients. The rest of the forms are rare. The progress of these diseases is devastating and includes progressive difficulties in speaking, dysphagia, dystonia, spasticity, parkinsonism, pure akinesia, oculomotor dysfunction, loss of vision and neuropsychiatric symptoms. Patients require the attention of a multidisciplinary team and they receive palliative treatment that is primarily focused on reducing dystonia and spasticity, which are highly disabling. In 2015, we initiated a project funded by Foundation La Marató of TV3, which is coordinated by Dr. B. Pérez-Dueñas (HU Vall d'Hebron). The aim of the project is to develop a clinical scale for PKAN patients, and to genetically diagnose the Spanish NBIA population with the collaboration of more than 20 Spanish hospitals. Some of the clinicians who are part of this network are part of the research team of this project. The PKAN-DSR clinical scale (PKAN Disease Rating Scale) has been successfully validated in the clinical evaluation of our series. Currently, we have registered 134 NBIA or NBIA-mimic cases, which refer to those forms of the disease that do not strictly show the clinical picture characteristic of these patients. The genetic analysis of PANK2 and PLA2G6 by Sanger sequencing and MLPA allowed us to diagnose 54% of the cases. Patients, who are negative for these analyses, are studied by means of a panel designed with the technology SureSelectXT (Agilent) for MiSeq (Illumina), which comprises 498 genes involved in NBIA and other movement disorders (MovDisord-498). 38 patients have been analysed with this methodology and so far, 19 of them have been genetically diagnosed. An example of this is the patient NBIA-18, a girl with consanguineous parents. Two homozygotic changes have been detected in two genes: DLD and FBXO7. We carried out studies of expression (western-blot), nonsense mediated decay (NMD) and subcellular localization in fibroblasts. These revealed that the FBXO7 c.368C>G mutation (p.S123X) leads to the activation of the NMD mechanism and mRNA is degraded; and that the DLD c.100A>G mutation (p.T34A) prevents the protein from reaching its correct location in the mitochondria. Negative cases after their study using the MovDisord-498 panel are studied by whole exome sequencing (WES). Approximately 85% of the pathological mutations are located in the exons. Therefore, although WES comprises approximately 2% of the human genome, it is an interesting cost-effective tool for genetic diagnosis. In pediatric neurodevelopmental diseases, comparative studies have shown that when WES is applied only to the proband sample, the success rate is around 28%, while if the study is extended to the parents (trio) the yield increases to 38%. It is important to emphasize that most cases are sporadic and, therefore, de novo mutations or mutations with an autosomal recessive inheritance are expected. We have studied two NBIA families (in both cases trios) with WES, and we have identified a novel gene not associated with any human pathology to date, and the second case of PLEKHG2 involved in leukodystrophy and acquired microcephaly with or without dystonia (MIM 616763). Undoubtedly, WES allows the characterization of new genes and new mutations in known genes, which permits a better characterization of the cl | es_ES |
| dc.format.extent | 50 | es_ES |
| dc.language | Español | es_ES |
| dc.publisher | Universitat Politècnica de València | es_ES |
| dc.rights | Reconocimiento - No comercial - Sin obra derivada (by-nc-nd) | es_ES |
| dc.subject | gen PLEKHG2 | es_ES |
| dc.subject | Atrofia cerebelar | es_ES |
| dc.subject | ENACH (Enfermedades Neurodegenerativas con Acumulación Cerebral de Hierro) | es_ES |
| dc.subject | Diagnóstico genético | es_ES |
| dc.subject | Exoma | es_ES |
| dc.subject | NGS (Next Generation Sequencing) | es_ES |
| dc.subject | Exome | es_ES |
| dc.subject | Genetic diagnosis | es_ES |
| dc.subject | NBIA (Neurodegeneration with Brain Iron Accumulation) | es_ES |
| dc.subject | Cerebellar atrophy | es_ES |
| dc.subject | PLEKHG2 gene | es_ES |
| dc.subject.classification | BIOQUIMICA Y BIOLOGIA MOLECULAR | es_ES |
| dc.subject.other | Grado en Biotecnología-Grau en Biotecnologia | es_ES |
| dc.title | Complejidad de las bases genéticas de los trastornos NBIA | es_ES |
| dc.type | Proyecto/Trabajo fin de carrera/grado | es_ES |
| dc.rights.accessRights | Abierto | es_ES |
| dc.contributor.affiliation | Universitat Politècnica de València. Departamento de Biotecnología - Departament de Biotecnologia | es_ES |
| dc.contributor.affiliation | Universitat Politècnica de València. Escuela Técnica Superior de Ingeniería Agronómica y del Medio Natural - Escola Tècnica Superior d'Enginyeria Agronòmica i del Medi Natural | es_ES |
| dc.description.bibliographicCitation | Guillot Fernández, M. (2019). Complejidad de las bases genéticas de los trastornos NBIA. http://hdl.handle.net/10251/127560 | es_ES |
| dc.description.accrualMethod | TFGM | es_ES |
| dc.relation.pasarela | TFGM\109950 | es_ES |
Este ítem aparece en la(s) siguiente(s) colección(ones)
-
ETSIAMN - Trabajos académicos [3278]
Escuela Técnica Superior de Ingeniería Agronómica y del Medio Natural

Universitat Politècnica de València. Unidad de Documentación Científica de la Biblioteca (+34) 96 387 70 85 · RiuNet@bib.upv.es
El contenido de este sitio está bajo una licencia Creative Commons Reconocimiento – No Comercial – Sin Obra Derivada (by-nc-nd), salvo que se indique lo contrario.
Los metadatos de este sitio están bajo una licencia Dominio Público.
