


De la genética a la función: análisis del efecto funcional de variantes genéticas con potencial efecto patológico en enfermedades neurodegenerativas
RiuNet: Repositorio Institucional de la Universidad Politécnica de Valencia
JavaScript is disabled for your browser. Some features of this site may not work without it.
Buscar en RiuNet
Listar
Mi cuenta
Estadísticas
Ayuda RiuNet
Admin. UPV

De la genética a la función: análisis del efecto funcional de variantes genéticas con potencial efecto patológico en enfermedades neurodegenerativas
Mostrar el registro sencillo del ítem
Ficheros en el ítem

| dc.contributor.advisor | Niñoles Rodenes, Regina
|
es_ES |
| dc.contributor.advisor | Pérez Tur, Jordi
|
es_ES |
| dc.contributor.author | Miravet Martí, Marc
|
es_ES |
| dc.date.accessioned | 2023-09-08T12:11:18Z | |
| dc.date.available | 2023-09-08T12:11:18Z | |
| dc.date.created | 2023-07-27 | |
| dc.date.issued | 2023-09-08 | es_ES |
| dc.identifier.uri | http://hdl.handle.net/10251/196099 | |
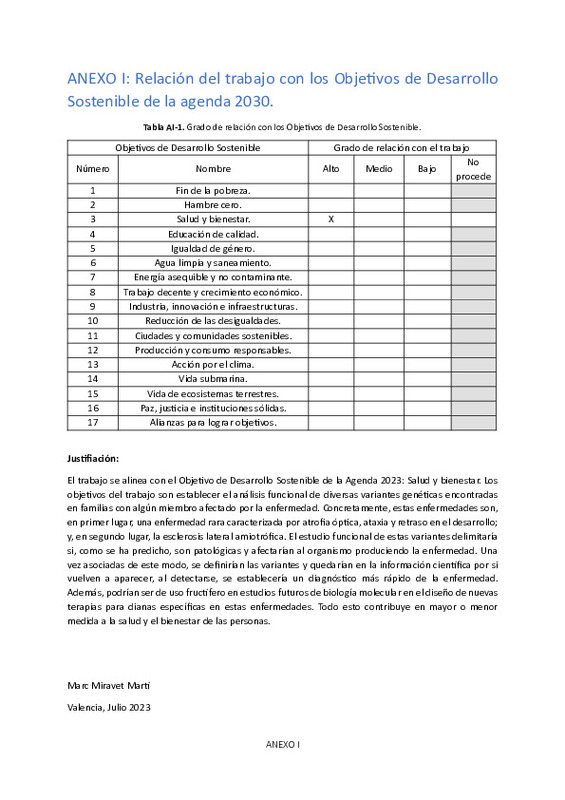
| dc.description.abstract | [ES] Una de las aproximaciones más exitosas a la hora de determinar los mecanismos moleculares implicados en la aparición de enfermedades humanas se encuentra en la genética molecular. La identificación de variantes con efectos patogénicos relacionadas con esas enfermedades suele servir como punto de partida para desgranar los mecanismos moleculares que entran en juego. Eventualmente, el conocimiento generado a partir de estos hallazgos genéticos puede dar lugar a aproximaciones terapéuticas con ciertas probabilidades de éxito. Sin embargo, el salto entre genotipo y fenotipo resulta muchas veces difícil de realizar. La mera identificación de variantes no implica comprender cómo juegan un papel, muchas veces determinante, en la aparición de los fenotipos de interés. El trabajo pretendió comenzar a explicar cómo ciertas variantes sospechosas de dar lugar a diversas enfermedades neurodegenerativas alteran la función de las proteínas en las que aparecen o de los genes a los que afectan. Se estudiaron dos enfermedades concretas, la primera: atrofia óptica 5 asociada a ataxia y retraso en el desarrollo, con variantes identificadas en DNM1L (Dynamin 1 Like), y en ACAD9 (Acyl-CoA Dehydrogenase Family Member 9). En segundo lugar, formas familiares de esclerosis lateral amiotrófica (ELA), relacionadas con variantes en KIF5A (Kinesin Family Member 5A), y SETX (Senataxin). En cuanto a la metodología, en OPA5, el análisis del efecto funcional se realizó utilizando fibroblastos obtenidos de los miembros de la familia, tanto de los pacientes de la enfermedad, portadores de variantes, y de sus padres, cada uno de los cuales es portador de una variante en cada uno de los genes. Como etapas iniciales del estudio, analizamos la expresión de estos genes mediante extracción de mRNA y RT-qPCR. Además, dado el papel de estos en la dinámica mitocondrial, mediante inmunocitoquímica y microscopía de fluorescencia, se intentó observarla. Por otro lado, se estudiaron las variantes observadas en KIF5A y en SETX, mediante la cuantificación de la expresión del mRNA a través de RT-qPCR y de la proteína del plasma sanguíneo de los individuos afectados y sanos, a través de Western blotting. Los resultados sugieren que existe una incorrecta oligomerización de DRP1 debido a su variante, que no se obtienen unos datos coherentes del estudio de la variante en ACAD9 y que la inmunocitoquímica de las estas proteínas tiene que ser mejorada. Sin embargo, todo apunta a que puede haber una sinergia clara que produzca la atrófia óptica. Mientras que las variantes en KIF5A, la intrónica es probablemente patogénica mientras que la missense sería benigna, y en SETX, la variante nonsense afecta en gran medida a la expresión y cantidad de proteína. Este trabajo se relaciona con el siguiente Objetivo de Desarrollo Sostenible (ODS) de la Agenda 2030: Salud y bienestar. | es_ES |
| dc.description.abstract | [EN] One of the most successful approaches to determining the molecular mechanisms involved in the development of human diseases lies in molecular genetics. The identification of variants with pathogenic effects related to these diseases often serves as a starting point for unraveling the molecular mechanisms at play. Eventually, the knowledge generated from these genetic findings can lead to therapeutic approaches with certain probabilities of success. However, bridging the gap between genotype and phenotype is often challenging to accomplish. The mere identification of variants does not imply understanding how they play a, often decisive, role in the occurrence of the phenotypes of interest. The work aimed to begin explaining how certain variants suspected of causing various neurodegenerative diseases alter the function of the proteins in which they appear or the genes they affect. Two specific diseases were studied: optic atrophy 5 associated with ataxia and developmental delay, with variants identified in DNM1L (Dynamin 1 Like) and ACAD9 (Acyl-CoA Dehydrogenase Family Member 9). Secondly, familial forms of amyotrophic lateral sclerosis (ALS) related to variants in KIF5A (Kinesin Family Member 5A) and SETX (Senataxin). Regarding the methodology, in OPA5, the analysis of functional effects was performed using fibroblasts obtained from family members, including disease patients carrying variants and their parents, each of whom is a carrier of a variant in each of the genes. As initial stages of the study, we analyzed the expression of these genes through mRNA extraction and RT-qPCR. Additionally, given their role in mitochondrial dynamics, attempts were made to observe it through immunocytochemistry and fluorescence microscopy. On the other hand, the observed variants in KIF5A and SETX were studied by quantifying mRNA expression through RT-qPCR and the plasma protein of affected and healthy individuals through Western blotting. The results suggest that there is incorrect oligomerization of DRP1 due to its variant. Coherent data from the study of the variant in ACAD9 and the immunocytochemistry of these proteins need to be improved. However, everything points to a clear synergy that produces optic atrophy. Regarding the variants in KIF5A, the intronic variant is likely pathogenic, while the missense variant would be benign. In SETX, the nonsense variant greatly affects protein expression and quantity. This work relates to the following Sustainable Development Goal (SDG) of the 2030 Agenda: Good health and well-being. | es_ES |
| dc.format.extent | 46 | es_ES |
| dc.language | Español | es_ES |
| dc.publisher | Universitat Politècnica de València | es_ES |
| dc.rights | Reserva de todos los derechos | es_ES |
| dc.subject | Análisis funcional | es_ES |
| dc.subject | Ataxia | es_ES |
| dc.subject | Esclerosis lateral amiotrófica | es_ES |
| dc.subject | OPA5 | es_ES |
| dc.subject | Variantes genéticas | es_ES |
| dc.subject | Amyotrophic lateral sclerosis | es_ES |
| dc.subject | Functional analysis | es_ES |
| dc.subject | Genetic variants | es_ES |
| dc.subject.classification | BIOQUIMICA Y BIOLOGIA MOLECULAR | es_ES |
| dc.subject.other | Grado en Biotecnología-Grau en Biotecnologia | es_ES |
| dc.title | De la genética a la función: análisis del efecto funcional de variantes genéticas con potencial efecto patológico en enfermedades neurodegenerativas | es_ES |
| dc.title.alternative | From genetics to function: analysis of the functional effect of genetic variants with potential pathological effects in neurodegenerative diseases | es_ES |
| dc.title.alternative | De la genètica a la funció: anàlisi de l efecte funcional de variants genètiques amb potencial efecte patològic en malalties neurodegeneratives | es_ES |
| dc.type | Proyecto/Trabajo fin de carrera/grado | es_ES |
| dc.rights.accessRights | Abierto | es_ES |
| dc.contributor.affiliation | Universitat Politècnica de València. Departamento de Biotecnología - Departament de Biotecnologia | es_ES |
| dc.contributor.affiliation | Universitat Politècnica de València. Escuela Técnica Superior de Ingeniería Agronómica y del Medio Natural - Escola Tècnica Superior d'Enginyeria Agronòmica i del Medi Natural | es_ES |
| dc.description.bibliographicCitation | Miravet Martí, M. (2023). De la genética a la función: análisis del efecto funcional de variantes genéticas con potencial efecto patológico en enfermedades neurodegenerativas. Universitat Politècnica de València. http://hdl.handle.net/10251/196099 | es_ES |
| dc.description.accrualMethod | TFGM | es_ES |
| dc.relation.pasarela | TFGM\156707 | es_ES |
Este ítem aparece en la(s) siguiente(s) colección(ones)
-
ETSIAMN - Trabajos académicos [3278]
Escuela Técnica Superior de Ingeniería Agronómica y del Medio Natural

Universitat Politècnica de València. Unidad de Documentación Científica de la Biblioteca (+34) 96 387 70 85 · RiuNet@bib.upv.es
El contenido de este sitio está bajo una licencia Creative Commons Reconocimiento – No Comercial – Sin Obra Derivada (by-nc-nd), salvo que se indique lo contrario.
Los metadatos de este sitio están bajo una licencia Dominio Público.
